
すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
小児および成人のアンジェルマン症候群
記事の医療専門家

最後に見直したもの: 04.07.2025

「体に気をつければ病気にはならない」といった言葉が、少なくとも馬鹿げているように聞こえる病気がいくつかあります。これらは、出生前から子供の体に何らかの精神的・身体的異常が内在しているものの、親の責任ではない病態です。こうした病気は染色体セットの変異や異常によって引き起こされ、染色体性疾患または遺伝性疾患と呼ばれます。アンジェルマン症候群、ダウン症候群、パトウ症候群、エドワーズ症候群、ターナー症候群、プラダー・ウィリー症候群など、これらはかなりまとまった遺伝性疾患のほんの一部に過ぎません。
ハッピーマン症候群
今回は、イギリスの小児科医ハリー・アンジェルマンにちなんで名付けられた病理についてお話します。アンジェルマンは1965年、この問題を初めて提起しました。前日、診療所で3人の変わった子供たちに遭遇し、彼らには共通の特異な症状が見られました。アンジェルマンはこれらの子供たちを「人形の子供たち」と呼び、彼らについて論文を書きました。当初、その論文は「マリオネットの子供たち」と題されていました。この論文とタイトルは、ヴェローナの美術館で見た絵画にインスピレーションを得て書かれたものです。その絵画は笑っている少年を描いており、「人形の少年」と呼ばれていました。絵画に描かれた子供と、アンジェルマンがかつて診療所で出会った3人の子供を関連付けたため、アンジェルマンは子供たちの病気を鑑みて、これらの子供たちを一つのグループにまとめました。
記事で言及された子供たちが他の医師の目に留まらなかったことは、驚くべきことではありません。一見すると、3症例とも全く異なる病気を抱えているように見えました。病状の全体的な臨床像が3症例とも大きく異なっていたからです。おそらくこの「新しい」染色体病理学は他の科学者の関心を引いたでしょうが、当時の遺伝学はまだイギリス人医師の仮説を裏付けるほどには発達していませんでした。そのため、ある程度の関心が寄せられた後、記事は長い間棚の奥にしまい込まれていました。
アンジェルマン症候群(イギリスの小児科医G・アンジェルマンの論文が現在アンジェルマン症候群と呼んでいる)に関する次の言及は、20世紀初頭に遡ります。そして1987年になってようやく、なぜ一部の子供たちが、外見上は常に笑顔で幸せそうに見えるような異常な状態で生まれるのか、その原因が解明されました。実際には、これは全く真実ではなく、その笑顔はただのしかめっ面であり、その裏には不幸な人間の魂と親の苦しみが隠されているのです。
疫学
統計によると、子供の染色体変異は、両親に同様の変異がある場合でも、またそのような変異がない場合でも発症する可能性があります。アンジェルマン症候群(AS)に明確な遺伝性はありませんが、染色体変異を持つ両親の場合、病態を発症する確率は非常に高くなります。
また、興味深いことに、家族にすでに AS の子供がいる場合、両親が健康であっても、同じ障害を持つ 2 番目の子供が生まれる可能性が 1 パーセントあります。
アンジェルマン症候群の患者数に関する正確な統計はまだありません。おそらく、症状の多様性が原因と考えられます。症状は特定の組み合わせで現れる場合もあれば、長期間全く現れない場合もあります。この疾患の有病率は、新生児20,000人あたり1人と推定されていますが、この数字は非常に概算です。
原因 アンジェルマン症候群
アンジェルマン症候群は染色体異常の医学的名称ですが、唯一の名称ではありません。この病気は人形児症候群、ハッピーパペット症候群、ペトルーシュカ症候群、笑い人形症候群などと呼ばれています。様々な呼び名が付けられ(患者本人やその親御さんにとって不快な名前さえあります)、どんなに奇妙に見えても、どんな理由があろうと、病気は病気です。
アンジェルマン症候群の発症原因は、他の多くの遺伝病と同様に、いずれの場合も染色体の1本、あるいは染色体セット全体の構造異常です。しかし、この症例では、問題は母親から受け継がれた15番染色体にあります。つまり、この症例では父親由来の染色体に異常はありませんが、女性の染色体に特定の変異が生じているのです。
アンジェルマン症候群は、染色体異常の種類に応じて染色体変異に分類されます。このような変異は、以下の通りです。
- 欠失(特定の遺伝子セットを含む染色体のセクションが欠如している状態。遺伝子の 1 つが欠落している場合は微小欠失と呼ばれます)は、2 回の切断と 1 回の再結合の結果であり、元の染色体のセクションが失われます。
- 重複(染色体内に既存の染色体のコピーである余分な部分が存在すること)は、ほとんどの場合に人の死につながり、まれに不妊症につながることもあります。
- 逆位(染色体のセクションの 1 つが 180 度反転、つまり反対方向に反転し、その中の遺伝子が逆の順序で配置されること)とは、染色体の破損した端が元の順序とは異なる順序で接続される場合です。
- 挿入(染色体中の遺伝物質の一部が本来あるべき位置から外れている場合)
- 転座(染色体の特定の部分が別の染色体に付着している場合。このような突然変異は、部分が失われることなく相互に起こり得る)。
何も知らない母親から変異した染色体を受け継いだ子供は、必ず異常を持って生まれます。アンジェルマン症候群の最も一般的な原因は、母親の15番染色体の欠失(小さな部分が欠損している状態)であると考えられています。「笑い人形」症候群における、あまり一般的ではない変異としては、以下のものが挙げられます。
- 転座、
- 片父性ダイソミー(子どもが父親から一対の染色体を受け継いだ場合、母親の染色体は存在しない)
- DNA 内の遺伝子の変異。DNA は、主要な構成要素 (遺伝物質) であると同時に、その正しい使用方法を指示する役割も担っています (特に、母親の染色体にある ube3a 遺伝子の変異)。
両親にこれらの変異のいずれかが存在する場合、小児におけるアンジェルマン症候群発症の危険因子となります。しかし、染色体変異だけでなく、ゲノム変異(染色体セットの量的変化を伴い、染色体変異よりも一般的)も、小児におけるアンジェルマン症候群の発症を引き起こす可能性があります。一般的なゲノム変異には、染色体トリソミー(染色体セットが46本を超える場合)が含まれます。
小児に病変が現れるには、両親が染色体異常を持っている必要は全くありません。しかし、遺伝性の疾患を持つ患者も一定数存在します。
病因
生物学、より正確には遺伝学についてもう少し深く掘り下げてみましょう。人間の個々の生物の遺伝情報は、23対の染色体に格納されています。各対の染色体の片方は父親から、もう片方は母親から子供に受け継がれます。すべての染色体対は形や大きさが異なり、特定の情報を持っています。したがって、23番目の染色体対(X染色体とY染色体)は、赤ちゃんの性的特徴(XXは女の子、XYは男の子、Y染色体は父親からのみ受け継がれます)の形成を担っています。
理想的には、子供は両親から46本の染色体を受け継ぎ、それらが遺伝的特徴を形成し、個人としての人格を決定づけます。染色体数が多い場合はトリソミーと呼ばれ、標準からの逸脱とみなされます。例えば、染色体セット(核型、種と個人の特徴を決定する)に47番染色体が存在すると、ダウン症候群が発生します。
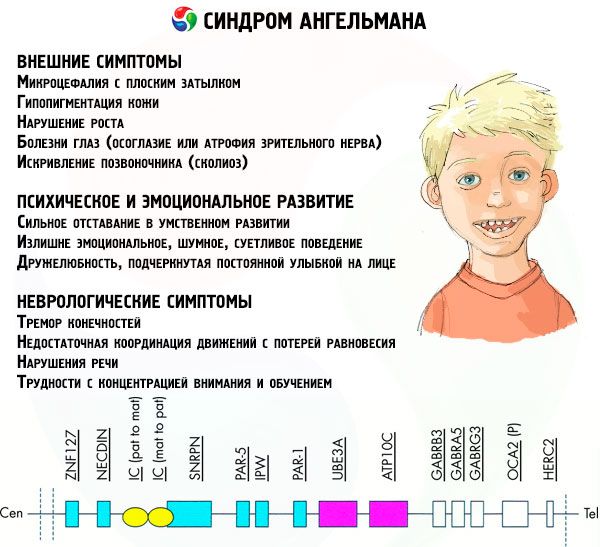
染色体を特殊な染料で染色すると、顕微鏡で見ると、それぞれの染色体に沿って異なる色合いの縞模様が見られます。それぞれの縞模様の中には、膨大な数の遺伝子が存在します。これらの縞模様はすべて科学者によって番号が付けられ、特定の位置にあります。縞模様の1つが欠落している場合、正常範囲からの逸脱とみなされます。アンジェルマン症候群では、母体染色体の長腕に位置するq11-q13領域(DNA塩基数は約400万)の欠落が非常に多く見られます。
染色体の主成分は、数千個の遺伝子と数千万個から数億個の窒素塩基を含む、非常に長いDNA分子であると考えられています。例えば、アンジェルマン症候群をはじめとする様々な疾患の発症に関与する15番染色体には、1200個の遺伝子と約1億個の塩基が含まれています。DNA分子の構造に何らかの異常があれば、将来生まれる子供の外見や発達に確実に影響を及ぼします。
遺伝子に含まれる遺伝情報は、タンパク質またはRNAに変換されます。このプロセスは遺伝子発現と呼ばれます。このようにして、親から受け継いだ遺伝情報は、形と内容の両方を受け取り、それぞれ固有の女性または男性の後継者に具現化されます。
アンジェルマン症候群など、非典型的な遺伝形式を伴う病理は数多く存在します。アンジェルマン症候群では、両親から受け継いだ遺伝子が対になった染色体の一部として、両親の固有の刻印を持ち、さまざまな形で症状が現れます。
アンジェルマン症候群は、ゲノムインプリンティングの顕著な例です。ゲノムインプリンティングでは、子どもの体内での遺伝子発現は、どちらの親からアレル(父親と母親から受け継いだ、対になった染色体の同一部位に位置する同一遺伝子の異なる形態)を受け継いだかに直接依存します。つまり、母親の染色体の異常のみがこの症候群の発症につながり、父親の染色体の変異や構造異常は全く異なる病態を引き起こします。
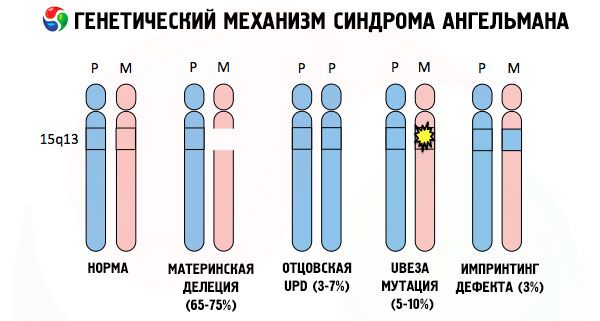
この病態では、母親の染色体における特定の遺伝子の欠損、または個々の遺伝子の活性の喪失/低下がみられます(多くの場合、他のタンパク質の分解を制御するタンパク質であるユビキチンの代謝に関与するube3a遺伝子が欠損しています)。その結果、子供は精神発達異常および身体変形と診断されます。
症状 アンジェルマン症候群
アンジェルマン症候群の症状は、子どもの生活と発達の様々な側面、すなわち身体的、神経学的、精神的側面に影響を及ぼします。このことから、この病状の進行を示す3つの症状群を特定することができます。
- 外的または身体的症状:
- 正常な大きさの体と手足に比べて不釣り合いに小さい頭、
- 口が広すぎる、
- 顔にはほぼ常に笑みが浮かんでいる(口を開けて)。
- 歯がまばらで、
- 上唇が狭い、
- 頻繁に舌を突き出す、
- 下顎が突き出ている、
- 尖った顎、
- 非常に白い肌、多くの場合髪の色(アルビノ、体内でメラニン色素が生成されないことと関連)
- 色白の肌に現れるシミ(メラニン生成不足による色素沈着低下)
- 身体的または外的症状:斜視や視神経萎縮などの眼疾患、
- 脊椎の湾曲(側弯症)
- 脚が硬い(歩くとき、関節の可動性が低いため、膝を曲げることができず、人形の歩き方に例えられる)。
- 精神的および感情的な発達に関連する症状:
- 重度の知的障害、
- 感情的すぎる、騒々しい、うるさい行動、
- 頻繁に手を叩く、
- 常に笑顔で親しみを表現し、
- 理由もなく頻繁に笑う。
- 神経症状:
- 手足の震え、
- バランスの喪失を伴う動作の不十分な調整、
- 筋緊張の低下、
- さまざまな睡眠障害、
- 幼少期に頻繁にヒステリー発作を起こした
- 言語障害(話し始めるのが遅く、コミュニケーション能力が低く、言葉が不明瞭である)
- 興奮性の増加を背景とした多動性、
- 集中力と学習能力の低下。
しかし、これはこの疾患の一般的なイメージです。実際には、アンジェルマン症候群の臨床像は、疾患の進行段階と病態を引き起こした染色体変異の種類に大きく依存します。つまり、この疾患の症状は患者ごとに大きく異なる可能性があり、長い間、この病態を同様の臨床像を持つ他の疾患と区別することができませんでした。
症状の総数の中で、例外なくすべての患者に共通する特徴的な症状を以下に挙げます。
- 重度の知的障害、
- 不適切な行動(不合理な笑い、興奮の増加、集中力の低下、陶酔状態)
- 運動能力の発達不全、
- 運動協調障害、歩行失調(不均一なペース、左右への揺れなど)、手足の震え。
- 非言語的コミュニケーション手段が優位となる言語発達障害。
大多数の患者が経験する症状の中には、次のようなものがあります。
- 身体の発達の遅れによって引き起こされる頭と体の不均衡、
- 多くの患者では頭蓋骨の形状により脳の大きさが健康な人よりも小さいままである(小頭症)。
- 3歳までにてんかん発作を起こし、年齢を重ねるにつれて発作の強さと頻度が徐々に減少する。
- EEG パラメータの歪み (低周波の変動と高振幅)。
これらの症状は非常に一般的ですが、アンジェルマン症候群の患者の 20% にはこれらの症状がありません。
さらに頻度は低いですが、次のような病気の症状を診断することも可能です。
- 重度または軽度の斜視、
- 舌の動きをうまく制御できず、患者が理由もなく舌を突き出すことが多くなる。
- 特に幼児の場合、嚥下や吸啜が困難である
- 皮膚と目の色素沈着の破壊、
- 歩行中に腕を上げたり曲げたり、
- 反射亢進、
- 睡眠障害、特に小児期、
- 頻繁な唾液分泌、
- 飽くことのない渇き、
- 過度に活発な咀嚼運動、
- 熱に対する過敏症、
- 頭の後ろが平らで、
- 下顎が突き出ている、
- 滑らかな手のひら。
患者の多くは排尿障害(排尿コントロールが困難)、微細運動能力の低下(セルフケアや学習の困難)、そして過体重といった問題を抱えています。ほぼすべての患者は、健常者よりも思春期が遅れて訪れます。
アンジェルマン症候群の子供は、口語を知覚し理解しますが、会話に参加したがらず、日常生活に必要な数十語程度しか話せません。しかし、成人期には、遺伝性疾患のない同年代の子供よりも若く見えます。
アンジェルマン症候群の症状の多くは不安定であるため、病状の臨床像は年齢とともに大きく変化します。けいれんやてんかん発作の頻度は減少するか、完全に消失し、患者の興奮性は低下し、睡眠は改善します。
合併症とその結果
アンジェルマン症候群は、重篤で現在事実上治癒不可能な染色体異常であり、患者から正常な生活を送る機会を奪います。アンジェルマン症候群の子どもの生活は、染色体異常の種類によって大きく左右されます。
染色体重複は、ほとんどの場合、生命維持に適しません。たとえ乳児期に死亡せず思春期を迎えたとしても、子供を持つ可能性はありません。
アンジェルマン症候群で最も多くみられる遺伝子の一部が欠失または欠損していることは、子どもが歩いたり話したりすることの習得を妨げます。このような子どもは、より重度の知的障害を呈し、てんかん発作の頻度と強度は、他の染色体異常を持つ患者よりもはるかに高くなります。
たった 1 つの遺伝子の変異であれば、適切な注意とアプローチで、子どもは自己管理、コミュニケーション、グループ内での交流の基礎を教えることができますが、それでも発達の面では同年代の子どもより遅れをとることになります。
アンジェルマン症候群の子どもたちは生まれつき優しいので、両親の愛情と関心が最も大切です。そうして初めて、たとえ小さな成果であっても、子どもたちの教育は実を結ぶのです。もちろん、アンジェルマン症候群の子どもたちは普通の学校では勉強できません。特別なクラスでまず集中力を教え、それから徐々に学校の基礎知識を身につけていく必要があります。
診断 アンジェルマン症候群
アンジェルマン症候群は先天性の発達障害です。しかし、特定の状況により、乳児期および幼児期には診断が不可能な場合が多くあります。これは、乳児および3歳未満の小児における症状の非特異性と弱い発現によるものです。また、我が国におけるこの疾患の有病率はそれほど高くないため、医師は同年代の患者と区別して認識するようになりました。
乳児におけるアンジェルマン症候群は、筋緊張の低下として現れることがあり、これは摂食障害(吸啜反射および嚥下反射の弱化)や、その後の歩行学習の困難(歩行開始がかなり遅れる)として現れます。これらの症状は、乳児の発達異常の最初の兆候であり、染色体異常に関連している可能性が高いです。この仮説を裏付けるには、遺伝子解析が必要です。
両親が様々なゲノム疾患や染色体疾患を抱えるお子さんには、特別な配慮が払われます。病気は初期には症状が現れない場合があり、早期に病状が判明すれば、お子さんと集中的に向き合うことで、学習の成功率が大幅に向上し、病気の進行を遅らせることができます。
両親にさまざまな染色体異常がある場合、SA は胎児段階で検出できる病状の 1 つであるため、赤ちゃんが生まれる前であっても遺伝子分析が行われます。
遺伝子研究のための材料の収集は、次の 2 つの方法で実行できます。
- 侵襲的(羊水サンプルを採取するために子宮を貫通する必要があるため、一定の割合のリスクを伴う)
- 非侵襲的(母親の血液から赤ちゃんの DNA を分析)。
次に以下の研究が行われます。
- 蛍光 in situ ハイブリダイゼーション(FISH 法) - 特殊な染料で標識された DNA プローブを研究対象の DNA に結合させ、顕微鏡で検査します。
- ube3a遺伝子およびインプリント遺伝子の変異の解析
- 遺伝学で使用される特殊な方法を使用した DNA メチル化分析。
遺伝子検査は染色体異常の場合、かなり正確な情報を提供してくれるため、将来の親は事前にどのような準備をすべきかを知ることができます。しかし、例外もあります。特定の患者群では、病理を示すすべての症状が見られるにもかかわらず、検査結果が正常のままとなることがあります。つまり、病理を特定するには、幼少期から子供を注意深く観察する必要があります。例えば、どのように食べるか、いつ歩き始め、いつ話し始めたか、歩くときに足を曲げるかどうかなどです。
アンジェルマン症候群の機器による診断方法としては、FISH法のほかに、脳の状態や大きさを調べるのに役立つ断層撮影法(CTまたはMRI)や、脳の個々の部分の働きを示す脳波(EEG)などがあります。
医師は通常、患者がすでにほとんどの症状を示し、病気の進行の経過が目に見えてわかる3~7歳で最終診断を下します。
どのようなテストが必要ですか?
差動診断
アンジェルマン症候群は、実質的に特異的な症状を示さない遺伝性疾患です。ほとんどの症状は、ASと他の遺伝性疾患の両方を示唆する可能性があります。
アンジェルマン症候群の鑑別診断は、以下の病状に基づいて行われます。
- ピット・ホプキンス症候群(患者は知的障害、明るい性格、笑顔、やや大きく広い口、小頭症を特徴とする)。違いは、覚醒時に過換気と息止めの発作を起こすことです。
- クリスチャンソン症候群(患者は、陽気な性格で話すことができない知的障害者で、小頭症、運動失調、けいれん、不随意筋運動を特徴とする)。
- モワット・ウィルソン症候群(症状:知的障害、てんかん発作、尖った顎、開いた口、幸福な表情、小頭症)。特徴:両眼の間隔が広い、目が内側に傾いている、鼻先が丸い、耳介が後ろ向きになっている。
- 歌舞伎症候群(軽度から中等度の知的障害、言語・運動障害、筋力低下、てんかん発作、小頭症、掻痒間隔の延長、協調運動障害を特徴とする)。アーチ状の眉毛、下眼瞼の外側部の外側への反り、離れ目、長い眼瞼裂、長く太いまつ毛が特徴です。
- レット症候群(女性におけるASとの鑑別)。症状:言語発達の遅れ、発作、小頭症。ASとの違いは、顔に喜びの表情がなく、無呼吸発作と失行発作が見られ、時間の経過とともに進行することです。
- 常染色体劣性精神遅滞症候群38(症状:運動能力および言語能力の遅れを伴う顕著な精神遅滞、筋力低下、乳児期の摂食障害、衝動性)。特徴的な特徴は虹彩の青色です。
- MECP2遺伝子重複症候群(男性におけるSAとの鑑別)。症状:重度の知的障害、幼少期からの筋力低下、言語障害または言語消失、てんかん。鑑別点:進行性ミオパチー、反復性感染症。
- クリーフストラ症候群(症状:言語・思考障害、筋力低下、睡眠障害、注意力の欠如、開口障害、多動性、発作、運動失調、平衡障害)。特徴的な特徴:平たい顔、短いしわしわの鼻、離れ目、大きく反り返った下唇、攻撃的な感情の爆発。
- スミス・マジェニス症候群(発作、睡眠障害、知的・運動発達障害を特徴とする)。特徴的な特徴として、幅広く平坦な顔と突き出た額が挙げられます。
- クーレン・ド・フリース症候群(軽度から中等度の知的障害、筋力低下、てんかん発作、友好的な性格)。特徴:長い顔と高い額、突き出た耳、つり上がった目、関節可動域の広さ、先天性心疾患。
- フェラン・マクダーミッド症候群(症状:知的障害、言語障害または言語消失)。特徴:筋肉が発達した大きな手、生まれつきの筋力低下、発汗量の減少。
アデニルコハク酸欠乏症、常染色体劣性精神遅滞症候群 1、染色体 2q23.1 重複症候群、FOXG1、STXBP1 または MEF2C 遺伝子ハプロ不全症候群などの病理は、アンジェルマン症候群に似た症状を「誇示」することがあります。
医師の仕事は、正確な診断を下し、アンジェルマン症候群を同様の症状を持つ病状と区別し、診断された病気の段階に適した効果的な治療を処方することです。
処理 アンジェルマン症候群
アンジェルマン症候群は、医学が未だに効果的な治療法を模索している病態の一つです。この疾患の病因的治療法は様々な方法や手段の開発段階にあり、その多くは未だヒトで試験されていません。そのため、医師は今のところ対症療法に頼らざるを得ず、てんかん発作、流涎、低血圧、睡眠障害といったマリオネット症候群の子供や大人のつらい状況を何とか緩和する治療に留まっています。
このように、適切に選択された抗てんかん薬の助けを借りれば、てんかん発作の頻度と強度を軽減することが可能です。しかし、SA患者の発作は、複数の種類の発作を特徴とする点で通常のてんかん発作とは異なり、複数の薬剤を同時に投与することで症状を緩和できるという点が大きな難点です。
アンジェルマン症候群の治療に最もよく用いられる抗てんかん薬は、バルプロ酸、トピラマート、ラモトリギン、レベチラセタム、クロナゼパム、およびこれらをベースとした薬剤です。カルマゼピン、フェニトイン、フェノバルビタール、エトスクシミドをベースとした薬剤はあまり使用されていません。これらの薬剤の中には、てんかん発作を増強し、頻度を増加させるという逆説的な作用を引き起こすものがあるためです。これは、これらの薬剤を単剤療法の一部として使用した場合に発生します。
よだれ症の治療には、通常、薬物療法(唾液の分泌を抑制する薬剤)と外科手術(唾液管の再移植)の2つの方法が用いられます。しかし、SAの場合、これらの方法は効果がないと考えられており、解決は未解決のままです。患者自身はよだれをコントロールできないことが多く、中には自分でよだれをコントロールできない人もいるため、親や介護者はこの問題に特に注意を払う必要があります。
もう一つの問題は睡眠時間の短さです。アンジェルマン症候群の子どもは5時間しか眠れないことが多く、これは全身の機能に悪影響を及ぼします。興奮しやすく活動的で、ゲームやコミュニケーション(たとえ非言語的な方法にとどめようと努力していても)が好きな子どもは、日中に明らかに疲れを感じます。十分な休息をとるためには、深く十分な睡眠が必要ですが、まさにこれが問題なのです。
興奮しやすい患者の睡眠を改善するには、神経系を落ち着かせる鎮静剤(フェノチアジン系薬剤や非定型抗精神病薬)で十分であるように思われます。しかし、ASの場合、これらの薬剤の使用は副作用を伴うことがあります。そのため、医師は依然として、メラトニン(睡眠ホルモンをベースとした天然ホルモン薬)やジフェンヒドラミンなどの穏やかな睡眠薬を好みます。メラトニンは就寝1時間前に1錠服用します。投与頻度と投与量は、患者の状態と年齢に応じて医師が決定します。
アンジェルマン症候群の患者さんは、消化や排便に問題を抱えることがあります。下剤(できればハーブ系のもの)を服用することで、便通を改善できます。
あるいは、アメリカの医師が行ったように、自閉症の治療法に基づいて、異なるアプローチで問題に取り組むこともできます。なぜなら、ASの特徴的な症状の多くは自閉症の特徴でもあるからです(衝動性、不随意運動、反復行動、注意欠陥、コミュニケーション障害など)。消化と排便を正常化するホルモンであるセクレチンの投与は、患者の注意力に良い影響を与え、オキシトシンは子供の認知能力と記憶力を向上させ、行動を矯正するのに役立つことが指摘されています。
確かに、ホルモン療法だけでは不十分です。特に子供の場合、なおさらです。アンジェルマン症候群では、行動療法、心理士、言語聴覚士(非言語コミュニケーション法と手話の指導)との連携が適応となります。このような子供たちの教育は、特別な訓練を受けた教師、心理士、そして両親の協力による個別プログラムに基づくべきです。残念ながら、これはどこでも可能なわけではなく、家族は問題を抱えたまま放置されてしまうのです。
ASの若年患者の多くは筋緊張低下や関節障害に悩まされているため、理学療法が重視されています。医師は多くの場合、パラフィン塗布、電気泳動、磁気療法などの治療法を用います。
積極的な強壮マッサージと特別な運動療法は、病気の子供がしばらくすると立ち上がって自信を持って歩けるようになるのに役立ちます。特に、冷水中でのアクア体操はSAに推奨されており、筋緊張を高め、子供の体のコントロールと動きの調整を促します。
抗けいれん薬による治療
アンジェルマン症候群の最も危険な症状は、てんかんに似た発作です。この症状は患者の80%に認められるため、すべての患者に効果的な抗てんかん薬による治療が必要です。
てんかん発作の治療は、ビタミン剤と抗てんかん薬を用いて行われます。けいれん症候群を伴うアンジェルマン症候群では、ビタミンB群に加え、ビタミンC、D、Eが効果的です。しかし、このような場合に自己判断でビタミン療法を行うことは非常に危険です。ビタミンの過剰摂取は抗てんかん薬の効果を低下させ、より重篤で持続的な新たな発作を引き起こす可能性があるからです。
抗てんかん薬の選択と有効な投与量の処方も専門医が行う必要があります。また、1種類の薬で十分なのか、それとも2種類以上の薬を長期間服用する必要があるのかについても、専門医が判断します。
ほとんどの患者に対して、医師は発作を予防し、患者の気分や精神状態を改善するバルプロ酸薬(バルプロ酸、デパキン、コンブレックス、バルパリンなど)を処方します。
バルプロ酸は、錠剤、シロップ、注射液の形で入手可能です。最も人気のある薬剤は、徐放性薬剤「デパキン」で、錠剤と静脈内投与用の溶液として提供されています。投与量は、患者の体重、年齢、状態に応じて医師が個別に決定します。
この薬は1日2~3回、食事中に服用します。1日の平均投与量は患者の体重1kgあたり20~30mg、最大投与量は1日あたり50mgです。
使用禁忌。肝機能障害、膵機能障害、出血性素因、肝炎、ポルフィリン症、または本剤に対する過敏症がある場合は使用しないでください。
副作用には、手の震え、消化および排便障害、体重の変化などがあります。
「トピラマート」もまた、SAの治療薬として選ばれています。錠剤として製造され、単剤療法として、また他の薬剤との併用療法として使用されます。
服用方法と用量。食事摂取の有無にかかわらず、錠剤を経口服用してください。成人の初期1日用量は25~50mg、小児は0.5~1mg/kgです。医師の指示に従って、1週間ごとに用量を増やしてください。
この薬は、妊娠中および授乳中、また成分に過敏症のある方は服用しないでください。この薬には様々な副作用があります。
アンジェルマン症候群に対して医師が処方する可能性のある薬:クロマゼパム、リボトリル、ラモトリギン、セイザール、ラミクタール、レベチラセタム、ケプラ、エピテラなど。
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
伝統医学とホメオパシー
ホメオパシー製剤のような伝統医学は、もちろん比較的安全ですが、アンジェルマン症候群に対するそのような治療法の有効性は議論の余地があると考えられます。
民間療法もいくつかの点で効果がありますが、ここではてんかん発作の抑制についてお話します。この点では、ハーブ療法は非常に効果的です。
シャクヤク、甘草、ウキクサをベースにした薬草コレクション(成分は同量ずつ摂取)は優れた効果をもたらします。これらのハーブは粉状に挽く必要があります。服用開始から2週間後には、発作の頻度が大幅に減少していることに気付くでしょう。
ラベンダーの煎じ薬(熱湯1杯につき小さじ1杯)も、生理痛に効果があります。5分間煮沸し、30分間浸出させます。14日間、毎晩服用してください。
マザーワートの水(またはアルコール)煎じ液はてんかん発作に効果があると考えられています。
アンジェルマン症候群の発作を予防するためのホメオパシー製剤としては、カモミールとマザーワート、アシダム・ヒドロシアニカム、アルゲンタム・ニトリカム、カリウム・ブロマタム、アルセニカム・アルブムをベースとした薬剤が挙げられます。ただし、個々の症例において効果的かつ安全な薬剤の投与量を処方できるのはホメオパシー医師のみであることをご承知おきください。
防止
読者の皆様は既にご存知かと思いますが、医学は遺伝子変異やその他の染色体異常を予防することも、その症状を治すこともまだできていません。アンジェルマン症候群の子どもは健康な両親から生まれており、遺伝学は医学の中でも最も研究が遅れている分野の一つであるため、この現象は誰にでも起こり得るのです。
唯一できることは、妊娠計画に責任あるアプローチを取り、登録を行い、適切な時期に検査を受けることです。しかし、繰り返しになりますが、こうした措置は、他の検査と同様に、予防というよりも教育的なものです。若い親は事前に何を準備すべきかを知っており、検査結果が陽性であれば、病気の子供を育てるという責任を引き受けられるかどうか判断するでしょう。
予測
アンジェルマン症候群の予後は、染色体異常の性質と、その発見の迅速さによって左右されます。最も深刻な影響を受けるのは、15番染色体に遺伝子の「ギャップ」(欠失)があるお子さんです。このようなお子さんが歩いたり話したりできるようになる可能性は極めて低いです。それ以外の場合は、お子さんへの丁寧なアプローチと愛情によって改善できる可能性があります。
残念ながら、このような患者は、決して愚かではなく、話し言葉とその意味を理解しているにもかかわらず、一人前の社会人になることはできません。しかし、生涯にわたってコミュニケーションに問題を抱えることになります。幼少期から手話を学ぶことはできますが、言葉でのコミュニケーションを強制することはできません。「話す」患者の語彙は、日常生活で使う最低限の言葉(5~15語)に限られています。
アンジェルマン症候群患者の平均余命と健康状態全般については、ここでの数値は平均値付近で変動しています。成人期には、脊柱側弯症や肥満といった健康問題に直面することが多くなりますが、適切な治療を受ければ生命を脅かすものではありません。