
すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
トリーチャー・コリンズ症候群
記事の医療専門家

最後に見直したもの: 04.07.2025

子宮内の骨の発育過程の障害は重篤な頭蓋顔面変形を引き起こし、こうした病状の 1 つにトリーチャー・コリンズ症候群 (TCS) または下顎筋膜、つまり顎顔面骨形成不全症があります。
ICD 10 による疾患コード:クラス XVII(先天異常、変形、染色体異常)、Q75.4 - 下顎顔面骨異形成症。
原因 トリーチャー・コリンズ症候群
この症候群は、100年以上前に病理の主要な特徴を記述した英国の著名な眼科医、エドワード・トリーチャー・コリンズにちなんで命名されました。しかし、ヨーロッパの医師は、この種の顔面骨および顎骨の異常を、スイスの眼科医アドルフ・フランチェッティによる広範な研究に基づいて、フランチェッティ病または症候群と呼ぶことが多いです。フランチェッティは、前世紀半ばに「下顎筋膜骨異形成症」という用語を導入しました。医学界では、フランチェッティ・コリンズ症候群という名称も使用されています。
トリーチャー・コリンズ症候群は、ヒト胎児の頭蓋顔面部の形成を担う核小体リン酸化タンパク質をコードするTCOF1遺伝子(染色体5q31.3-33.3座)の変異によって引き起こされます。このタンパク質の量が早期に減少することで、rRNAの生合成と機能が阻害されます。ヒトゲノム研究プログラムの遺伝学者によると、これらのプロセスは、神経堤(神経溝に沿った隆起部で、胚発生中に神経管に閉じる)の胚細胞の増殖を減少させます。
顔面組織の形成は、神経堤上部(頭部)の細胞の形質転換と分化によって起こり、神経管に沿って胎児の第1鰓弓と第2鰓弓の領域まで移動します。これらの細胞の欠乏は、頭蓋顔面の変形を引き起こします。異常発生の臨界期は受精後18~28日です。神経堤細胞の移動が完了すると(妊娠4週目)、顔面領域のほぼすべての遊離間葉系組織が形成され、その後(5~8週目)、顔、首、喉頭、耳(内耳を含む)、そして将来の歯のすべての部分の骨格組織と結合組織に分化します。
病因
トリーチャー・コリンズ症候群の病因は多くの場合家族性で、常染色体優性遺伝で異常が遺伝しますが、常染色体劣性遺伝で異常が遺伝するケースもあります(他の遺伝子、特にPOLR1CおよびPOLR1Dの変異を伴う)。顎顔面骨異形成症において最も予測困難な点は、変異が子供に遺伝するのは症例の40~48%に過ぎないことです。つまり、患者の52~60%では、トリーチャー・コリンズ症候群の原因は家族内の異常の存在とは関連しておらず、散発的な遺伝子変異の結果として病理が発生すると考えられています。新しい変異は、妊娠中の胎児への催奇形性影響の結果である可能性が高いです。
この症候群の催奇形性原因として、専門家は大量のエタノール(エチルアルコール)、放射線、タバコの煙、サイトメガウイルス、トキソプラズマ、グリホサート系除草剤(ラウンダル、グリフォー、トルネードなど)を挙げています。また、医原性因子としては、13-シス-レチノイン酸を含むニキビ治療薬や脂漏症治療薬(イソトレチノイン、アキュテイン)、抗てんかん薬フェニトイン(ジランチン、エパヌチン)、向精神薬ジアゼパム、バリウム、レラニウム、セドクセンなどが挙げられます。
症状 トリーチャー・コリンズ症候群
下顎筋膜異骨症の臨床症状とその発現の程度は、ほとんどの場合、遺伝子変異の発現特性に依存します。そして、この異常の最初の兆候は、ほとんどの場合、出生直後の乳児に現れます。トリーチャー・コリンズ症候群の顔面は特徴的な外観を呈します。さらに、形態学的異常は通常、両側性かつ対称的です。
トリーチャーコリンズ症候群の最も明らかな症状は次のとおりです。
- 頭蓋骨の顔面骨の発育不全(低形成):頬骨、前頭骨の頬骨突起、外側翼突板、副鼻腔、下顎および骨端線(顆)の突出。
- 下顎骨の発達不全(小顎症)および通常よりも鈍角な下顎角。
- 鼻は正常な大きさであるが、側頭領域の頬骨弓の形成不全および頬骨弓の発達不全または欠損のために大きく見える。
- 目の切れ込みが下向きになっている、つまり目の形が異常で、目尻が下向きに垂れ下がっている。
- 下まぶたの欠損(コロボーマ)および下まぶたのまつ毛の部分的な欠損。
- 耳介の形状が不規則で、下顎の角に位置する、耳介葉がない、耳珠と口角の間に盲瘻があるなど、さまざまな偏差がある。
- 外耳道の狭窄または閉鎖(閉鎖)および中耳耳小骨の異常。
- 耳下腺唾液腺の欠如または低形成;
- 咽頭低形成(咽頭と気道の狭窄)
- 硬口蓋の癒合不全(口蓋裂)、および軟口蓋の欠如、短縮、または不動状態。
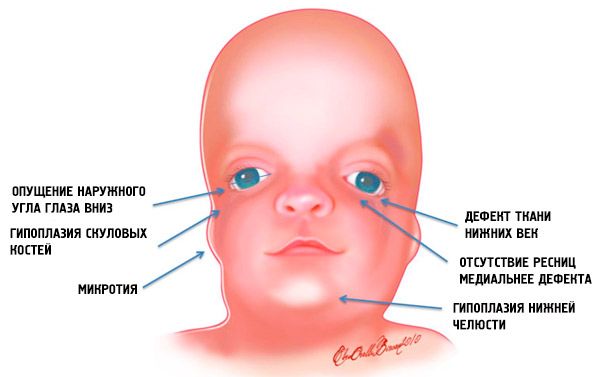
このような解剖学的異常は、いずれの場合も合併症を伴います。具体的には、伝音難聴または完全難聴といった機能的聴覚障害、眼球形成不全による視覚障害、口蓋欠損による摂食・嚥下障害などが挙げられます。顎欠損に伴う歯列不正(不正咬合)は、咀嚼や発音に問題を引き起こします。軟口蓋の病変は、鼻声の原因となります。
診断 トリーチャー・コリンズ症候群
トリーチャー・コリンズ症候群の出生後診断は、基本的に臨床症状に基づいて行われます。頭蓋顔面骨異形成症は、症候群が完全に発現している場合は容易に診断できますが、病理学的症状が軽微に発現している場合は、正しい診断を確立することが困難になる可能性があります。
この場合、異常に関連するすべての機能、特に呼吸に影響を与える機能(睡眠時無呼吸のリスクがあるため)の評価に特別な注意を払う必要があります。また、栄養補給の有効性とヘモグロビン酸素飽和度も評価・モニタリングする必要があります。
その後、出産後5~6日目に、産科病院で行われる聴覚検査によって聴覚障害の程度を判断する必要があります。
検査が処方され、その際に頭蓋顔面異形学の透視検査、パントモグラフィー(顔面頭蓋骨の骨構造のパノラマX線)、さまざまな投影による全頭蓋コンピューター断層撮影、内耳道の状態を判断するための脳のCTまたはMRIによる機器診断が行われます。
家族歴にトリーチャー・コリンズ症候群がある場合、顎顔面異常の最も早期の(出生前の)診断は、妊娠10~11週の絨毛生検によって可能です(この処置は流産や子宮の感染の恐れがあります)。
家族からも血液検査が行われます。妊娠16~17週目には羊水が分析され(経腹的羊水穿刺)、妊娠18~20週目には胎児鏡検査が行われ、胎盤の胎児血管から血液が採取されます。
しかし、ほとんどの場合、超音波は胎児のこの症候群の出生前診断(妊娠20〜24週)に使用されます。
どのようなテストが必要ですか?
処理 トリーチャー・コリンズ症候群
遺伝的に決定された先天性欠損症の他の症例と同様に、重症型のトリーチャー・コリンズ症候群の治療は、このような病態に対する治療法が全く存在しないため、対症療法のみとなります。この症候群における変形の範囲と程度は広範であるため、医療介入の性質と強度にも多くの選択肢があります。
補聴器は聴力を矯正し改善するために使用され、言語療法セッションは発話を改善するために使用されます。
重度の気道狭窄(気管切開)や喉頭狭窄(栄養補給のため胃瘻造設)の場合は、早期に外科的介入が必要となります。また、口蓋の外科的矯正が必要になる場合もあります。
下顎骨延長手術は2~3歳以降に行われます。軟部組織再建には、下眼瞼コロボーマ矯正術や耳介形成術が含まれます。
予測
この病状の予後はどのようなものでしょうか?変形の程度と症状の強さによって異なります。トリーチャー・コリンズ症候群は生涯にわたる診断です。
 [ 25 ]
[ 25 ]